Structure and usage of clusters
This page is adapted from my Crystal Growth project notebook with added contents, which was written between Feb. and Mar. 2022 and shared within the group for induction and training proposes. Both this page and the notebook are under regular inspection, but their contents are not synced.
Divide a job: Nodes, Processors and Threads
Node: A bunch of CPUs and probably with GPUs / coprocessors for acceleration. Memory and input files are shared by processors in the same node, so a node can be considered as an independent computer. The communication between nodes are achieved by ultra-fast network, which is the bottleneck of modern clusters.
Processor: The unit to deal with a ‘process’, also known as ‘central processing unit’, or CPU. Processors in the same node communicate via shared memory.
Thread: Subdivision of a process. Multiple threads in the same process share the resources allocated to the CPU.
The figure below illustrates the hierarchy of node, processor, and thread:
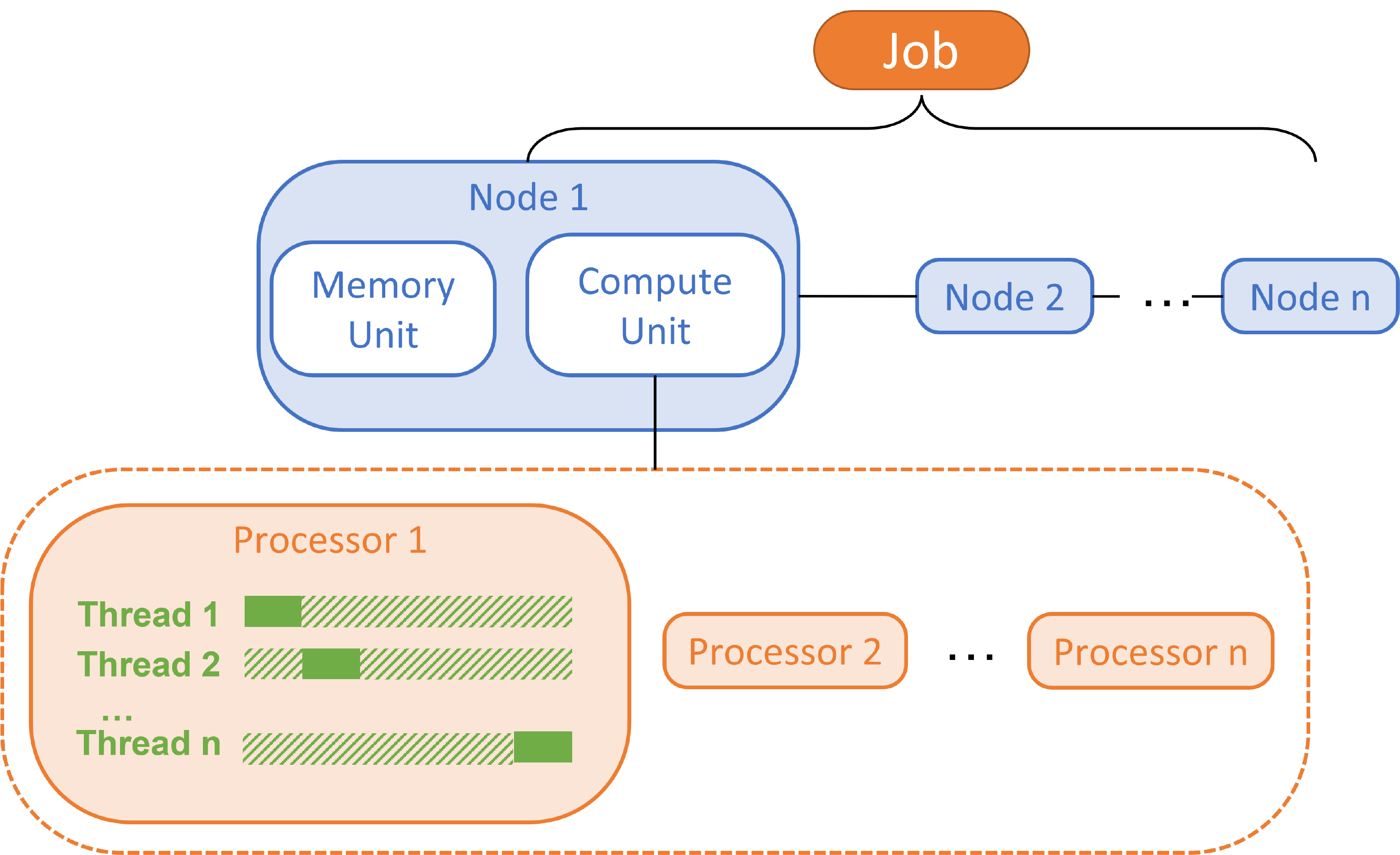
Multiple processes vs multiple threads
From the figure above, it is not difficult to distinguish the differences between a ‘process’ and a ‘thread’: process is the smallest unit for resource allocation; thread is part of a process. The idea of ‘thread’ is introduced to address the huge difference in the speed of CPU and RAM. CPU is always several orders of magnitude faster than RAM, so typically the bottleneck of a process is loading the required environment from RAM, rather than computations in CPU. By using multiple threads in the same process, various branches of the same program can be executed simultaneously. Therefore, the shared environmental requirements doesn’t need to be read from RAM for multiple times, and the loading time for threads is much smaller than for processes.
However, multithreading is not always advantageous. A technical prerequisite is that the program should be developed for multithread proposes. Python, for example, is a pseudo-multithread language, while Java is a real one. Sometimes multithreading can lead to catastrophic results. Since threads share the same resource allocation (CPU, RAM, I/O, etc.), when a thread fails, the whole process fails as well. Comparatively, in multiple processes, other processes will be protected if a process fails.
In practice, users can either run each process in serial (i.e., number of threads = 1), or in parallel (i.e., number of threads > 1) on clusters. However, the former is recommended, because of more secured resource managements. The latter is not advantageous. Besides the problem mentioned above, it might lead to problems such as memory leak when running programs either: not developed for multithreading / requires improper packages (See the famous libfabric issue on ARCHER2) - common problems of scientific computing packages & their users. :-)
More nodes vs more CPUs
When the allocated memory permits, from my experience, using more CPUs/processes per node is usually a better idea, considering that all nodes have independent memory space and the inter-node communications are achieved by wired networks. It almost always takes longer to coordinate nodes than to coordinate processors within the same node.
The internal coordinator: What is MPI
Message passing interface, or MPI, is a standard for communicating and transferring data between nodes and therefore distributed memories. It is utilized via MPI libraries. The most popular implementations include:
- MPICH - an open-source library;
- Intel MPI - a popular implementation of MPICH especially optimised for Intel CPUs;
- OpenMPI - an open-source library;
- OpenMP - Not MPI; parallelization based on shared memory, so only implemented in a single node; can be used for multithreading;
In practice, a hybrid parallelization combining MPI and OpenMP to run multithread jobs on cluster is allowed, though sometimes not recommended. The first process (probably not a node or a processor) is usually allocated for I/O, and the rest is used for parallel computing.
So far, MPI only supports C/C++ and FORTRAN, which explains why all parallel computing software is based on these languages. To launch an executable in parallel, one should specify: mpiexec or mpirun.
Technical details about how MPI is implemented is too advanced for typical computational chemists / solid state physicists, which is a specific, completely different research area, so they are not covered in this page.
Distribute files: The role of job submission scripts
As mentioned in previous sections, memories are distributed to nodes. Considering the inefficiency of inter-node communication, it is unpractical to frequently transfer files across nodes. In practice, required input files are duplicated, or synced, to all the nodes before any calculation. During the calculation, data is transferred from computing nodes between the I/O node.
Although such sync is automatic for almost any modern cluster, it is safer to specify that when developing job submission scripts:
1
$ ssh ${NODEADDRESS} "${COMMAND}"
Secure your storage: Work directory and home directory
All the modern clusters have separate disk spaces for differently proposes, namely, work directory and home directory. This originates again from the famous speed difference between CPU and RAM/ROM. 2 distinctly kinds of disks are used respectively to improve the overall efficiency and secure important data:
- For work directory, large, high-frequency disks are used. Data stored in work directory is usually not backed up, and in some cases, will be automatically cleaned after a fixed time length.
- For home directory, mechanical disks with slower read/write frequency but better robustness are used. Usually files in home space are backed up.
For large clusters like ARCHER2, the work directory and the home directory are completely separated, i.e., directory is only viable by login nodes; work directory is viable by both job and login nodes. Job submission in home directory is prohibited.
For more flexible, medium-sized clusters like Imperial CX1, submitting jobs in home directory and visiting home directory by job nodes are allowed, yet storing temporary files during calculation in home directory is still not recommended because of the potential influence on other files and the reduced overall efficiency. Work and home directories can be called with the following environmental variables:
${EPHEMERAL} - Work directory. Data will be cleaned over 30 days.
${HOME} - Home directory.
Setup your environment: What does an application need?
Executable
The binary executable should, theoretically, all be stored in \usr\bin. This never happens in practice, unless you are a fanatical fundamentalist of the early Linux releases. To guide your system to the desired executable, you can either laboriously type its absolute path every time you need it or add the path to the environmental variable:
1
$ export PATH=${PATH}:path_to_bin
Running any executable in parallel requires mpi to coordinate all the processes/threads. The path to mpi executable is also required. Besides, many scientific codes require other specific environmental variables such as linear algebra packages. Read their documentations for further information.
lib/a/o files
When writing a script, you might need some extra packages to do more complex jobs. Those packages are developed by experts in computer science and can be called by a line of code. The same thing happens when people were developing applications like CRYSTAL and ONETEP.
However, scientific computing codes are usually distributed in the form of source code. Source codes in FORTRAN/C/C++ need be compiled into a binary executable. There are 2 options during compiling:
- Include the whole package as long as one of its functions is called, also known as a ‘static lib’.
- Only include a ‘table of contents’ when compiling, also known as ‘dynamic lib’. The packages needed are separately stored in ‘dll/so’ files, making it possible for multiple applications sharing the same lib.
Details about compilation are beyond the scope of this post. Maybe I will make another post to discuss them, when I am more confident with this topic. The thing is: when running a dynamically linked application, information should be given to help the code find the libs needed. This can be specified by:
1
$ export LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:path_to_lib`
For statically linked applications, usually you need not worry about it - but the volume of the compiled executable might make you feel wonder whether there is an alternative way.
Conflicts
Improper previous settings may lead to a wrong application, or a wrong version, if multiple applications with similar functions are installed in the system, such as Intel compiler and GCC, OpenMPI and MPICH - a common phenomenon for shared computing resources. To avoid this, the path to the undesired application or lib should be removed from the environmental variables.
Environmental Modules
Environmental modules is a popular software managing the necessary environmental setups and conflicts for each application. It can easily add or erase the environmental variables by commands module load or module rm, and modulefiles written in Tool Command Language (TCL). The default directory of modulefiles is given in the environmental variable ${MODULEPATH}, but files in other directories can also be loaded by their absolute path.
Both Imperial CX1 and ARCHER2 adopt this application, which pre-compiled applications offered.
The external coordinator: What is a batch system
Always bear in mind that the computational resources are limited, so you need to acquire reasonable resources for your job. Besides, the cluster also needs to calculate your budget, coordinate jobs submitted by various users, and make the best of available resources. When job is running, maybe you also want to check its status. All of this are fulfilled by batch systems.
In practice, a Linux shell script is needed. Parameters of the batch system of are set in the commented lines at the top of the file. After the user submit the script to batch system, the system will:
- Examine the parameters
- Allocate and coordinate the requested resources
- Set up the environments, such as environmental variables, package dependency, and sync the same setting to all nodes
- Launch a parallel calculation - see mpi part
- Post-process
Note that a ‘walltime’ is usually required for a batch job, i.e., the maximum allowed time for the running job. The job will be ‘killed’, or suspended, when the time exceeds the walltime, and the rest part of the script will not be executed. timeout command can be used to set another walltime for a specific command.
Common batch systems include PBS, which involves different releases, and Slurm. For Imperial cluster CX1 and MMM Hub Young (managed by UCL), PBS system is implemented; for ARCHER2 and Tianhe-2 LvLiang(天河二号-吕梁), Slurm is implemented. Tutorials of batch systems are not covered here, since they are highly machine-dependent - usually modifications are made to enhance the efficiency. Refer to the specific user documentations for more information.
How to run a job in parallel: Things to consider
Successfully setting and submitting a batch job script symbolizes that you do not need this tutorial any more. Before being able to do that, some considerations might be important:
- How large is my system? Is it efficient to use the resources I requested(Note that it is not a linear-scaling problem… Refer to this test on CRYSTAL17)?
- To which queue should I submit my job? Is it too long/not applicable/not available?
- Is it safe to use multi-threading?
- Is it memory,GPU etc. demanding?
- Roughly how long will it take?
- What is my budget code? Do I have enough resources?
- Which MPI release version is my code compatible with? Should I load a module or set variables?
- Any other specific environmental setups does my code need?
- Do I have any post-processing script after MPI part is finished? How long does it take?